-

上海沙格医疗科技有限公司
主营:FDA认证,FDA注册,欧盟授权代表,fda510k认证,MDRCE认证

上海沙格医疗科技有限公司
主营:FDA认证,FDA注册,欧盟授权代表,fda510k认证,MDRCE认证 7
7

FDA认可机构,前FDA官员全程,FDA注册,510k,美代,*,价格优
医疗器械FDA 510(k)注册,因其相应FD&C Act*510,故通常称510(K)注册。510(k)为医疗器械在美国上市的主要途径之一,绝大多数的II类医疗器械和部分I类、III类医疗器械通过此途径清关上市。对510(k)注册文件所必须包含的信息,FDA有一个基本的要求,其内容大致如下16个方面:
1) 申请函,此部分应包括申请人(或联系人)和企业的基本信息、510(K)递交的目的、申请上市器械的名称型号和分类资料、进行实质等效比较的产品(Predicate Device)名称及其510(k)号码;
2) 目录,即510(k)文件中所含全部资料的清单(包括附件);
3) 真实性保证声明,对此声明,FDA有一个标准的样本;
4) 器材名称,即产品通用名、FDA分类名、产品贸易名;
5) 注册号码,如企业在递交510(k)时已进行企业注册,则应给出注册信息,若未注册,也予;
6) 分类,即产品的分类组、类别、管理号和产品代码;
7) 性能标准,产品所满足的强制性标准或自愿性标准;
8) 产品标识,包括企业包装标识、使用说明书、包装附件、产品标示等;
9) 实质相等性比较(SE);
10) 510(k)摘要或声明;
11) 产品描述,包括产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等;
12) 产品的*性与有效性,包括各种设计、测试资料;
13) 生物相容性;
14) 色素添加剂(如适用);
15) 软件验证(如适用);
16) 灭菌(如适用),包括灭菌方法的描述、灭菌验证产品包装和标识等。
FDA510K 流程
实质相等性比较(SE)
实质相等性比较是要所申请上市的产品和已在美国市场上合法销售的产品在*性和有效性方面比较是实质相等的。选择合适的产品进行比较是510(K)申请的关键步骤。在进行比较时应从如下方面进行考虑:
企业必须提供充足的资料,所申请上市的器械和被比较的器械是实质相等的(SE),否则510(k)申请不会通过。
510(K)审查程序
FDA在收到企业递交的510(k)资料后,先检查资料是否齐全,如资料齐全,则受理并给企业发出确认性,同时给出申请受理编号(K YYXXXX),此号码也将作为正式批准后的号码;如不齐全,则要求企业在规定时间内补充齐全,否则作企业放弃处理。FDA在受理申请后即进入内部工作程序,其中可能还会要求企业补充一些资料。在510(k)申请通过审阅后,FDA并不立即发出批准函件,而是根据产品风险等级、市场先前是否对企业有不良反 映等确定是否对企业进行现场GMP考核,考核通过后再发给企业正式批准函件(Clearance);如无须现场考核GMP,则510(k)申请通过后立即发给正式批准函件。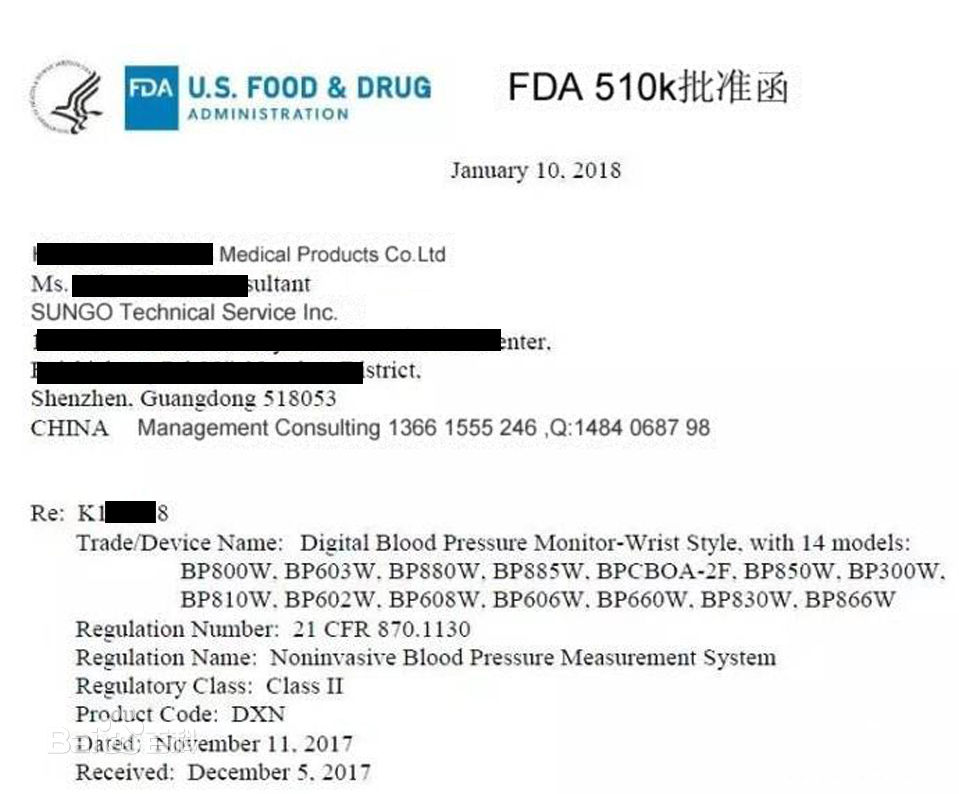
根据风险程度,FDA将医疗器械分为三类(I,II,III),风险.FDA明确规定了其产品分类和管理要求对于每种设备,目前有**过1,700种FDA医疗器械目录。任何想要进入美国市场的设备必须先确定分类和管理要求要列出的产品的rements。
对于任何产品,企业都需进行企业注册(Registration)和产品列名(Listing)
对Ⅰ类产品(占47%左右),实行的是一般控制(General Control),绝大部分产品只需进行注册、列名和实施GMP规范,产品即可进入美国市场(其中少数产品连GMP也豁免,少数保留产品则需向FDA递交510(K)申请即PMN(Premarket Notification))
对Ⅱ类产品(占46%左右),实行的是控制(Special Control),企业在进行注册和列名后,还需实施GMP和递交510(K)申请(少产品是510(K)豁免)
对Ⅲ类产品(占7%左右),实施的是上市前许可,企业在进行注册和列名后,须实施GMP并向FDA递交PMA(Premarket Application)申请(部分Ⅲ类产品还是PMN
对Ⅰ类产品,企业向FDA递交相关资料后,FDA只进行公告,并无相关发给企业;对Ⅱ、Ⅲ类器械,企业须递交PMN或PMA,FDA在公告的同时,会给企业以正式的市场准入批准函件(Clearance),即允许企业以自己的名义在美国医疗器械市场上直接销售其产品。至于申请过程中是否到企业进行现场GMP考核,则由FDA根据产品风险等级、管理要求和市场反馈等综合因素决定
综合以上内容可知,绝大部分产品在进行企业注册、产品列名和实施GMP,或再递交510(K)申请后,即可获得FDA批准上市
FDA 的职责是确保美国本国生产或进口的食品、化妆品、药物、生物制剂、设备和放射产品的*
上述产品,包括“医疗器械”,必须经过FDA认证后,方可在美国市场上销售
医疗器械的FDA认证,有如下几种
厂家在FDA注册
产品的FDA登记
产品上市登记
产品上市审核批准
其他
根据风险等级的不同,FDA将医疗器械分为三类(Ⅰ,Ⅱ,Ⅲ),Ⅲ类风险等级
少量I、III类,多数II类的医疗器械,在美国销售,需要做“产品上市登记”(PMN : Premarket Notification)的认证
做“产品上市登记”所需提交的文件需满足美国法规 FD&C Act*510。
通常称做“产品上市登记”这类的认证为510(K)认证
对Ⅰ类产品,企业向FDA递交相关资料后,FDA只进行公告,并无相关发给企业;对Ⅱ、Ⅲ类器械,企业须递交PMN或PMA,FDA在公告的同时,会给企业以正式的市场准入批准函件(Clearance),即允许企业以自己的名义在美国医疗器械市场上直接销售其产品。至于申请过程中是否到企业进行现场GMP考核,则由FDA根据产品风险等级、管理要求和市场反馈等综合因素决定。
综合以上内容可知,绝大部分产品在进行企业注册、产品列名和实施GMP,或再递交510(K)申请后,即可获得FDA批准上市
FDA510(k)申报、文件编写服务
CE认证、*四版评估报告编写、MDR文件编写、MDR转版咨询、欧盟代表1366电1555话246
医疗器械FDA认证分类
医疗器械申请FDA的分类比较简单,就是I类和II类,III类,只是在I类和II类里,有些产品比较,所以在实际的FDA认证代理工作中,我们还遇到一些的类别,那就是在I类里,有些产品需要做510K,在II类产品里,有些可以豁免510K。所以,我们在实际工作中的划分方式为:
I类
II类
III类
I类不豁免510K
II类可豁免510K
医疗器械的认证周期要根据产品的类别来确定,一般I类产品,并且是豁免510K的话,这种比较快,15个工作日内就可以完成产品注册,在2009年10月份之前,3-5个工作日就可以取得注册码,2009年10月份以后,必须先支付费用,才能得到注册码,所以在支付费用的环节上,比原来严格了一点,以前是先给注册码,后支付费用,现在是先支付费用,后给注册码。这样的话,周期就延长了一点。如果不是豁免510K的产品,那么做510K的时间相对比较长,这个还要看客户选择哪个认证机构来审核510K报告,如果选择第三方机构来审核,时间相对快一点,一般3个月内基本可以拿到510K号码,如果选择FDA直接来审核,那就慢了,没有半年基本都不可能。
一、510K指的是FDA法规里面的一个,讲的是PMN,也就是Pre Market Notification 上市前通告。
510(k)文件是向FDA递交的上市前申请文件,目的是申请上市的器械与不受上市前批准(PMA)影响的合法上市器械同样*有效,即为等价器械(substantially equivalent)。申请者必须把申请上市的器械与现在美国市场上一种或多种相似器械对比,得出并且支持等价器械的结论。合法上市器械是在1976年5月28日之前合法上市的器械(preamendment device),或者从III类器械中分入II或I类的器械,或者通过(510(k))程序发现与这样的器械等价的器械,或者通过自动的III 类器械定义的评价建立的器械。与之等价的器械被称为“predicate device(s)”。
二、医疗器械FDA认证流程
一类医疗器械,并且是豁免510K的产品,流程比较简单,只要申请企业注册和产品注册即可,没什么复杂的。
II类医疗器械,并且不豁免510K的产品,流程稍微复杂一点,先要准备产品技术资料、产品工艺资料、产品参数资料、产品结构资料、产品测试数据等。有了这些资料,我们就可以撰写510K报告。撰写好510K报告,就要提交给FDA或第三方机构审核,审核通过后,就会取得一个510K代码,有了这个代码,就可以申请产品注册了,工厂注册随时可以申请。
大致流程就是:准备资料--撰写510K报告--510K报告提交机构审核--取得510K代码--产品注册和工厂注册--取得产品注册码和工厂注册码。
三、医疗器械FDA所需资料
对于I类豁免510K的医疗器械,做FDA注册相对比较简单,只要提供申请人信息(包括公司名称、地址、电话、联系人、邮箱、网站等)和产品英文名称即可,要是能多提供一些产品信息自然更好.
对于II类医疗器械,撰写FDA510K报告需要准备的资料比较多,大致的内容如下:
(1) 申请函,此部分应包括申请人(或联系人)和企业的基本信息、510(K)递交的目的、申请上市器械的名称型号和分类资料、进行实质等效比较的产品(Predicate Device)名称及其510(K)号码;
(2) 目录,即510(K)文件中所含全部资料的清单(包括附件)
(3) 真实性保证声明,对此声明,FDA有一个标准的样本;
(4) 器材名称,即产品通用名、FDA分类名、产品贸易名;
(5) 注册号码,如企业在递交510(K)时已进行企业注册,则应给出注册信息,若未注册,也予;
(6) 分类,即产品的分类组、类别、管理号和产品代码;(7) 性能标准,产品所满足的强制性标准或自愿性标准;
(8) 产品标识,包括企业包装标识、使用说明书、包装附件、产品标示等;
(9) 实质相等性比较(SE);
(10) 510(K)摘要或声明;
(11) 产品描述,包括产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等;
(12) 产品的*性与有效性,包括各种设计、测试资料;
(13) 常规测试项目: 生物相容性;产品性能。
(14) 色素添加剂(如适用);
(15) 软件验证(如适用);
(16) 灭菌(如适用),包括灭菌方法的描述、灭菌验证产品包装和标识等。
一、510K指的是FDA法规里面的一个,讲的是PMN,也就是Pre Market Notification 上市前通告。
510(k)文件是向FDA递交的上市前申请文件,目的是申请上市的器械与不受上市前批准(PMA)影响的合法上市器械同样*有效,即为等价器械(substantially equivalent)。申请者必须把申请上市的器械与现在美国市场上一种或多种相似器械对比,得出并且支持等价器械的结论。
二、医疗器械FDA认证流程
一类医疗器械,并且是豁免510K的产品,流程比较简单,只要申请企业注册和产品注册即可,没什么复杂的。
II类医疗器械,并且不豁免510K的产品,流程稍微复杂一点,先要准备产品技术资料、产品工艺资料、产品参数资料、产品结构资料、产品测试数据等。有了这些资料,我们就可以撰写510K报告。撰写好510K报告,就要提交给FDA或第三方机构审核,审核通过后,就会取得一个510K代码,有了这个代码,就可以申请产品注册了,工厂注册随时可以申请。
大致流程就是:准备资料--撰写510K报告--510K报告提交机构审核--取得510K代码--产品注册和工厂注册--取得产品注册码和工厂注册码。
三、医疗器械FDA所需资料
对于I类豁免510K的医疗器械,做FDA注册相对比较简单,只要提供申请人信息(包括公司名称、地址、电话、联系人、邮箱、网站等)和产品英文名称即可,要是能多提供一些产品信息自然更好.
对于II类医疗器械,撰写FDA510K报告需要准备的资料比较多,大致的内容如下:
(1) 申请函,此部分应包括申请人(或联系人)和企业的基本信息、510(K)递交的目的、申请上市器械的名称型号和分类资料、进行实质等效比较的产品(Predicate Device)名称及其510(K)号码;
(2) 目录,即510(K)文件中所含全部资料的清单(包括附件)
(3) 真实性保证声明,对此声明,FDA有一个标准的样本;
(4) 器材名称,即产品通用名、FDA分类名、产品贸易名;
(5) 注册号码,如企业在递交510(K)时已进行企业注册,则应给出注册信息,若未注册,也予;
(6) 分类,即产品的分类组、类别、管理号和产品代码;(7) 性能标准,产品所满足的强制性标准或自愿性标准;
(8) 产品标识,包括企业包装标识、使用说明书、包装附件、产品标示等;
(9) 实质相等性比较(SE);
(10) 510(K)摘要或声明;
(11) 产品描述,包括产品的预期用途、工作原理、动力来源、零组件、照片、工艺图、装配图、结构示意图等;
(12) 产品的*性与有效性,包括各种设计、测试资料;
(13) 常规测试项目: 生物相容性;产品性能。
(14) 色素添加剂(如适用);
(15) 软件验证(如适用);
(16) 灭菌(如适用),包括灭菌方法的描述、灭菌验证产品包装和标识等。
四、FDA510K评审周期
Day 1:FDA receives 510(K) submission
↓
By Day 7
FDA sends Acknowledgement Letter.
OR
FDA sends Hold Letter if unresolved issues with User Fee and/or eCpoy.
↓
By Day 15:
FDA conducts Acceptance Review.
FDA informs submitter if 510(K) is accepted for Substantive Review or placed on RTA Hold.
↓
By Day 60:
FDA conducts Substantive Review.
FDA communicates via a Substantive Interaction to inform the sunmitter that the FDA will either proceed with Ineractive Review or that 510(K) will be placed on hold and Additional Information is required.
↓
By Day 90:
FDA sends final MDUFA Decision on 510(K).
↓
By Day 100:
If MDUFA Decision is not reached by Day 100,FDA Provides Missed MDUFA Decision Communication that identifines outstanding review issues.
我司可以提供FDA 510(k)咨询的服务:
申请前策略咨询
根据产品特点,选择合适的对比器械
为您制定申请的解决方案
建立测试方案,提供测试服务
协助您准备510(k)文件所需的各类相关信息
为您编写510(k)文件
代表您与的审核机构进行沟通
协助您完成工厂注册和医疗器械列示
http://fdasungo.b2b168.com
欢迎来到上海沙格医疗科技有限公司网站, 具体地址是上海市浦东新区上海市浦东新区世纪大道1500号14楼,老板是张小姐173-2126-1042。
主要经营上海沙格医疗科技有限公司(电话:*)致力于为**的生产商和经营者提供市场准入的合规咨询以及注册服务。服务包含:FDA认证、FDA注册、ISO13485认证、欧盟授权代表、fda510k认证、MDRCE认证。十多年里,SUNGO已为**30多家上市公司和**制造商,合计5000多家企业提供过相关服务。
。
单位注册资金单位注册资金人民币 500 - 1000 万元。
你有什么需要?我们都可以帮你一一解决!我们公司主要的特色服务是:FDA认证,FDA注册,欧盟授权代表,fda510k认证,MDRCE认证等,“诚信”是我们立足之本,“创新”是我们生存之源,“便捷”是我们努力的方向,用户的满意是我们较大的收益、用户的信赖是我们较大的成果。